
-
Argentina

-
Australia

-
Austria

-
Bahrain (العربية)

-
Bahrain (English)
-
België (Nederlands)

-
Belgique (Français)
-
Brazil

-
Canada (English)

-
Canada (Français)
-
Chile

-
Colombia
-
Croatia

-
Denmark

-
Deutschland

-
Europe

-
France

-
Greece (Ελληνικά)

-
Italia

-
Hungary

-
Lietuva

-
México

-
日本 (日本語)

-
대한민국 (한국어)

-
Kuwait (العربية)

-
Kuwait (English)
-
Nederland

-
Norge

-
Oman (العربية)

-
Oman (English)
-
Polska

-
Portugal

-
Qatar (العربية)

-
Qatar (English)
-
Saudi Arabia (العربية)

-
Saudi Arabia (English)
-
Slovakia (Slovak)

-
Slovenia (Slovenščina)

-
Spain

-
Suomi

-
Sverige

-
Schweiz

-
台灣 (中文)

-
United States

-
UAE (العربية)

-
UAE (English)

 Genética
Genética
Confirmación genética para el diagnóstico de la Atrofia Muscular Espinal (AME)
Para asegurar el diagnóstico de Atrofia Muscular Espinal (AME), es necesaria la confirmación genética.
Tras la sospecha clínica de Atrofia Muscular Espinal (AME), el paciente debe ser remitido a una prueba genética para su diagnóstico, pues es un padecimiento de origen genético 1. El examen se realiza mediante la extracción de sangre, saliva o raspado de mejilla para la obtención de ADN y el análisis cuantitativo del gen SMN1.
En la actualidad, estas pruebas se realizan mediante las técnicas de biología molecular: ligación múltiple dependiente de amplificación de la sonda o MLPA por sus siglas en inglés (Multiplex Ligation-Dependent Probe Amplification), reacción en cadena de la polimerasa cuantitativa o qPCR por sus siglas en inglés (quantitative Polymerase Chain Reaction) o secuenciación de siguiente generación o NGS por sus siglas en inglés (Next Generation Sequencing).
El algoritmo diagnóstico se muestra en la siguiente figura1. Aunque algunos análisis también proporcionan la cuantificación del número de copias del gen SMN2, estos datos no son necesarios para el diagnóstico, pero son importantes para ayudar a predecir la severidad de la enfermedad1,2.
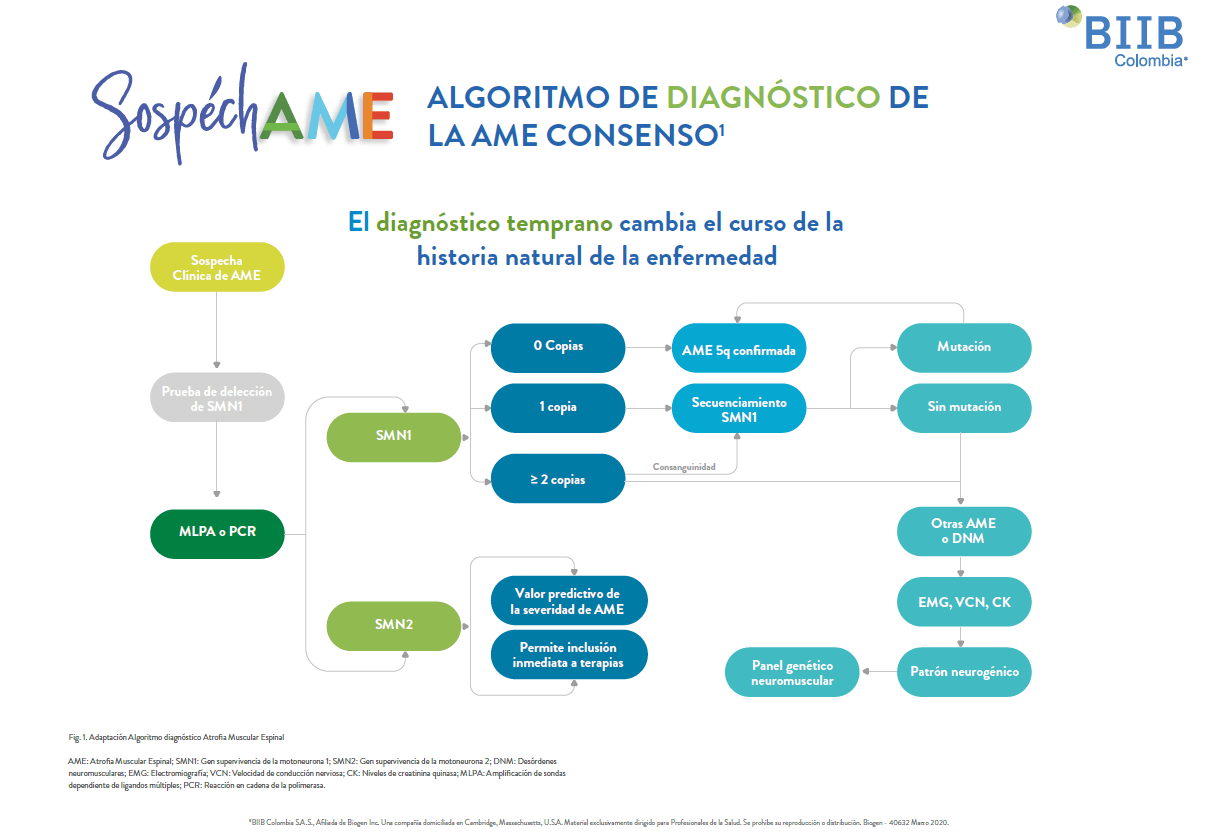

Diagnóstico de pacientes con AME en etapa presintomática por antecedente familiar.
Parejas que han tenido descendencia con AME tienen un 25% de probabilidades de tener hijos con la enfermedad. Por esta razón, es fundamental que los hermanos de los pacientes sean evaluados para AME1.
El diagnóstico de Atrofia Muscular Espinal (AME) puede realizarse al nacimiento o incluso en periodo prenatal, con la misma prueba genética que se realiza para los pacientes sintomáticos1.
El diagnóstico prenatal permite realizar intervenciones farmacológicas (si es necesario) antes del desarrollo de síntomas, lo cual mejora el pronóstico3. Además, es esencial que la familia sea referida a un asesoramiento genético.
El impacto del diagnóstico en el individuo con AME y su familia
El momento del diagnóstico es difícil para la familia y, por lo tanto, debe ser tratado con cuidado por los profesionales de la salud. Toda la expectativa que genera la llegada de un nuevo miembro a la familia se ve profundamente sacudida por el diagnóstico de una enfermedad grave, que a menudo llega tardíamente.
En este momento de fragilidad, es fundamental que la conducta de los profesionales sea empática y sensible, al proporcionar un entorno acogedor y de apoyo desde el principio, pero sin omitir la comunicación y la correcta información con la familia y según el grupo de edad, de los pacientes.
El impacto emocional y psicológico es considerable, por lo que el apoyo del paciente y su familia desde el primer momento puede tener un impacto positivo en su calidad de vida a largo plazo.
Referencias
1. Mercuri E, Finkel RS, Muntoni F, et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord. 2018;28(2):103-115.
2. Calucho M, Bernal S, Alías L, et al. Correlation between SMA type and SMN2 copy number revisited: An analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscul Disord. 2018;28(3):208-215.
3. Tizzano EF, Zafeiriou D. Prenatal aspects in spinal muscular atrophy: From early detection to early presymptomatic intervention. Eur J Paediatr Neurol. 2018;22(6):944-950.
4. Ministerio de Salud. Humaniza SUS. 2003. p. 1–19. Disponible en: http://www.saude.gov.br/acoes-e-programas/humanizasus
¿Tienes dudas sobre algún término de este artículo? Consulta el glosario.
#AtrofiaMuscularEspinal #Diagnóstico #Prueba genética #ConfirmaciónGenética #Genes #ADN