
-
Argentina

-
Australia

-
Austria

-
Bahrain (العربية)

-
Bahrain (English)
-
België (Nederlands)

-
Belgique (Français)
-
Brazil

-
Canada (English)

-
Canada (Français)
-
Chile

-
Colombia
-
Croatia

-
Denmark

-
Deutschland

-
Europe

-
France

-
Greece (Ελληνικά)

-
Italia

-
Hungary

-
Lietuva

-
México

-
日本 (日本語)

-
대한민국 (한국어)

-
Kuwait (العربية)

-
Kuwait (English)
-
Nederland

-
Norge

-
Oman (العربية)

-
Oman (English)
-
Polska

-
Portugal

-
Qatar (العربية)

-
Qatar (English)
-
Saudi Arabia (العربية)

-
Saudi Arabia (English)
-
Slovakia (Slovak)

-
Slovenia (Slovenščina)

-
Spain

-
Suomi

-
Sverige

-
Schweiz

-
台灣 (中文)

-
United States

-
UAE (العربية)

-
UAE (English)

 Sobre la AME
Sobre la AME
Manifestaciones clínicas de la Atrofia Muscular Espinal (AME)
Signos y síntomas de la Atrofia Muscular Espinal (AME).
Cada persona puede presentar diferentes síntomas y niveles de complicación. Queremos acompañarte para que conozcas más mientras lees aquí.
La Atrofia Muscular Espinal (AME) se subdivide clínicamente en cinco categorías, definidas por el número de copias del gen SMN2, la edad de inicio de los síntomas y las habilidades motoras alcanzadas. Cada niño puede presentar diferente sintomatología y con diferentes niveles de complicación9,10.
Es importante destacar que la información que presentamos se basa en la evolución de la enfermedad (historia natural), según los datos publicados en la literatura científica de los últimos años.
La posibilidad de una atención multidisciplinaria temprana, así como el desarrollo de nuevas terapias, podrían alterar significativamente la evolución de las manifestaciones de la AME10.
Atrofia Muscular Espinal (AME) Tipo 0
Severa (0-1 copia del gen SMN2)
Es la forma más compleja de AME y amenazante de la vida. Comienza en el periodo prenatal y además de la afectación motora y respiratoria, los pacientes con AME severa, también conocida como tipo 0, pueden presentar alteraciones cardíacas y cerebrales1,3.
Algunas de las dificultades que pueden presentar estos pacientes son:
Dificultades respiratorias: A menudo necesitan asistencia respiratoria en los primeros minutos u horas después del nacimiento.
Dificultades motoras: Hipotonía profunda, debilidad grave y contracturas articulares.
Dificultades de alimentación: Presentan disfagia grave e incapacidad para succionar.
Esperanza de vida: La gran mayoría de los pacientes fallecen poco tiempo después de la aparición de los síntomas.
Atrofia Muscular Espinal (AME) Tipo 1
Aguda Severa (1-2 copias del gen SMN2)
También conocida como enfermedad de Werdnig-Hoffman o tipo I, es el subtipo más común de AME, y corresponde a alrededor del 60% de los casos notificados en la literatura. Los signos y síntomas comienzan antes de los seis meses de vida1,4,5.
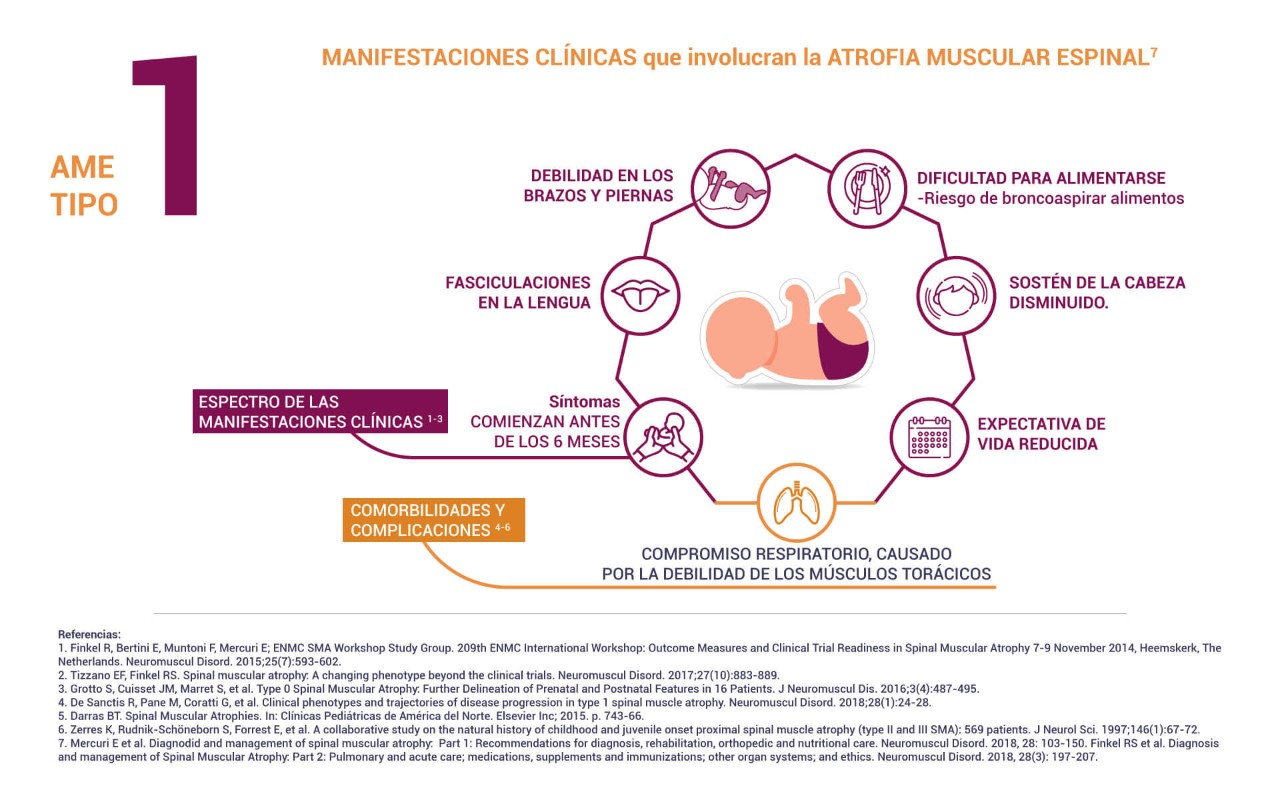

Algunas de las dificultades que pueden presentar estos pacientes son:
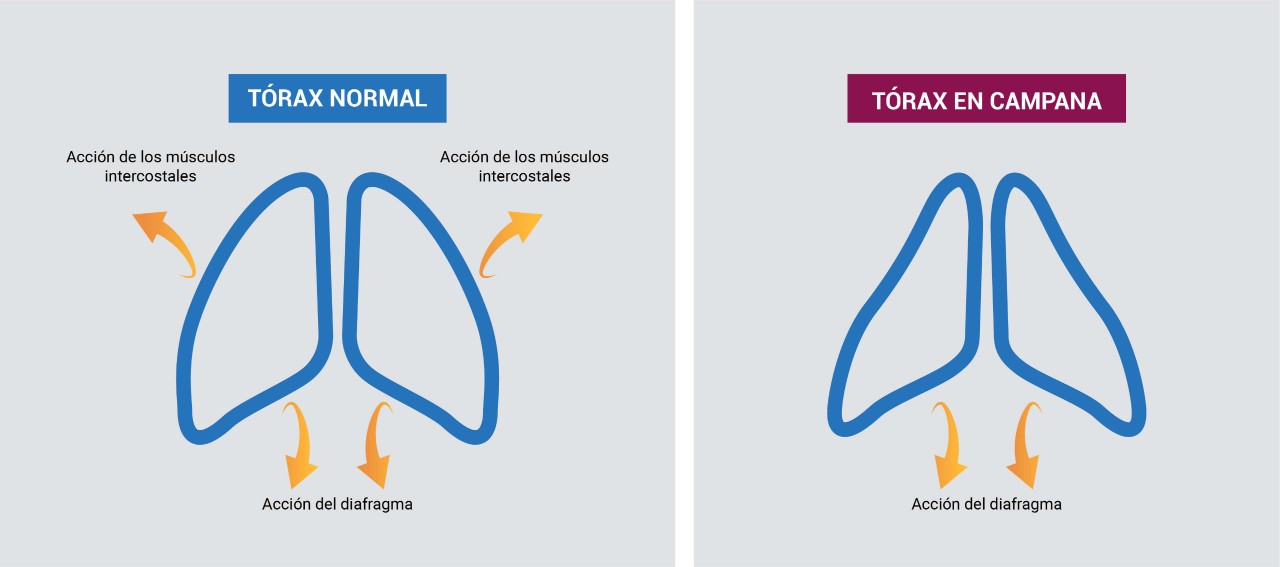
Dificultades respiratorias: Desarrollan una respiración paradójica e insuficiencia respiratoria que es la principal causa de morbilidad y mortalidad. La debilidad e hipotonía de los músculos respiratorios también causan alteraciones en el pecho, produciendo el llamado “tórax en forma de campana”.

Dificultades motoras: No desarrollan la capacidad de sentarse sin apoyo y tienen gran pérdida de movimiento en el primer año de vida.
Dificultades de alimentación: La afectación de los músculos de la lengua y la faringe provoca pérdida de la capacidad de succión y disfagia, lo que puede causar deficiencia nutricional y riesgo de bronconeumonías a repetición. Los niños con AME severa pueden necesitar soporte nutricional a través de un tubo gástrico. La afectación en la musculatura bulbar es responsable de fasciculaciones en la lengua (movimientos rápidos e involuntarios), además los pacientes con AME pueden presentar constipación grave.
Esperanza de vida: Sin tratamiento, lamentablemente, alrededor del 68% de los pacientes mueren antes de los dos años de edad, y el 84% antes de los cuatro años de edad. La adopción de cuidados respiratorios y nutricionales proactivos puede reducir la mortalidad antes de 2 años al 30%.
Atrofia Muscular Espinal (AME) Tipo 2
Crónica - Severa (2-3 copias del gen SMN2)
La AME crónica-severa también conocida como AME tipo II o síndrome de Dubowitz, es la forma intermedia de la enfermedad en la que los síntomas suelen comenzar entre los seis y los dieciocho meses de edad. Con una estimación del 29% de los casos reportados en la literatura1,6,7.
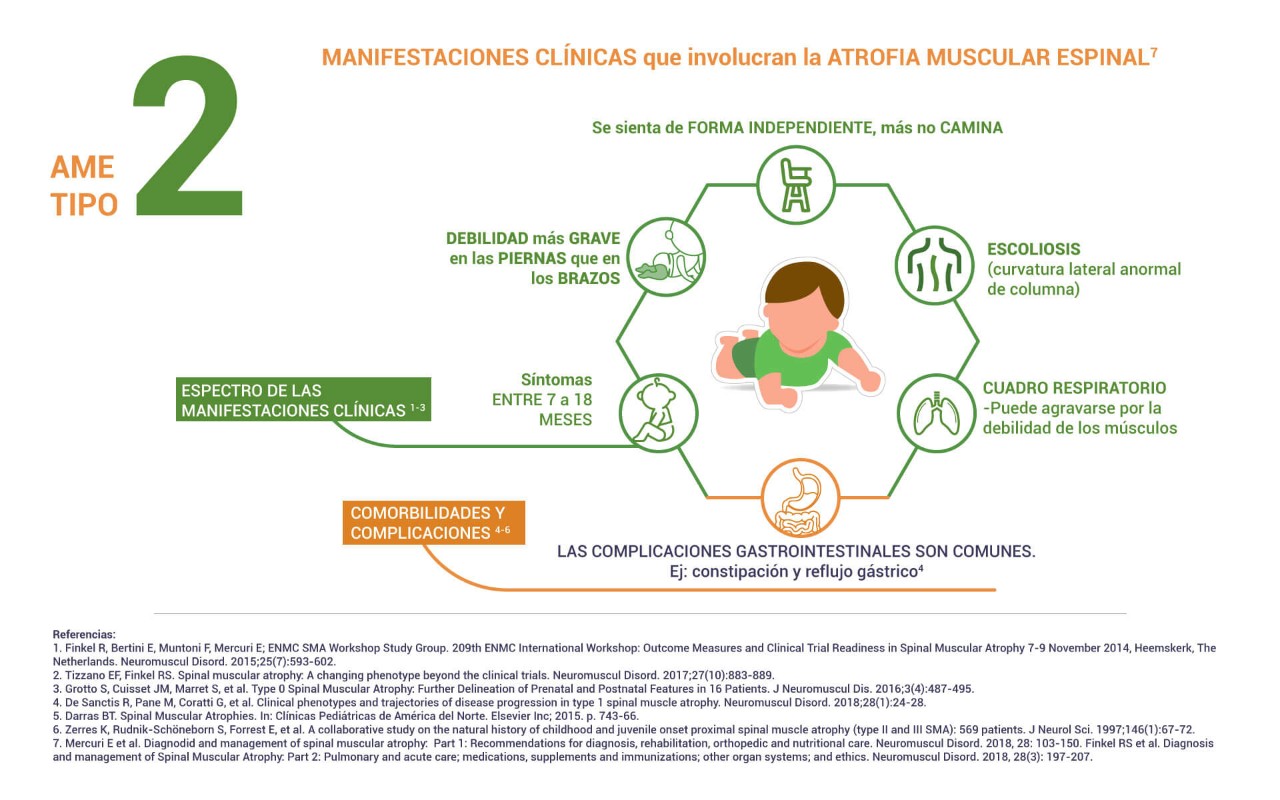

Algunas de las dificultades que pueden presentar estos pacientes son:
Dificultades respiratorias:Los pacientes pueden desarrollar hipoventilación, inicialmente durante el sueño, lo que requiere el uso de soporte ventilatorio nocturno, además de maniobras de fisioterapia para la correcta eliminación de secreciones.
Dificultades motoras: Las personas con AME crónica-severa pueden tener la capacidad de sentarse sin apoyo, pero pueden perder esta capacidad a medida que la enfermedad progresa. Algunos pacientes pueden estar de pie, pero no caminar de forma independiente. Muchos pacientes presentan contracturas y deformidades articulares que incluyen escoliosis severa.
Dificultades de alimentación: Con la progresión de la enfermedad, pueden desarrollar disfagia y debilidad bulbar y por ende, fallas en la deglución.
Esperanza de vida: Los estudios de historia natural muestran que los pacientes con AME crónica-severa llegan a la edad adulta, sin embargo, muestran una mortalidad temprana en contraste con la población general.
Atrofia Muscular Espinal (AME) Tipo 3
Moderada (2-3 copias del gen SMN2)
También conocida como AME tipo III o enfermedad de Kugelberg-Welander, afecta cerca del 13% de los casos. Los primeros síntomas aparecen después de los 18 meses de edad1,6.


Algunas de las dificultades que pueden presentar estos pacientes son:
Dificultades respiratorias: Algunos pacientes desarrollan dificultades respiratorias más tarde, en comparación con AME crónica-severa.
Dificultades motoras: Consiguen desarrollar la capacidad de caminar independientemente, pero en algún momento de la vida pueden perder esta capacidad. Cuanto empiezan a presentarse los signos y síntomas, rápidamente se puede perder de la marcha. Las dificultades óseas que incluyen la escoliosis, aumentan cuando se pierde la capacidad de caminar.
Dificultades de alimentación: En los casos más graves o con más tiempo de enfermedad, pueden desarrollar dificultades de deglución.
Esperanza de vida: Los estudios muestran que la esperanza de vida de estos pacientes es muy parecida en relación con la de la población sin AME.
Atrofia Muscular Espinal (AME) Tipo
Leve (>3 copias del gen SMN2)
La AME leve, también conocida como tipo 4, también es una de las formas más escasa de la enfermedad y representa menos del 5% de los casos. La mayoría de las veces, los primeros síntomas aparecen a partir de la segunda o tercera década de vida. La mayoría de las personas con AME leve no presentan dificultades para respirar o alimentarse1,9.
Dificultades motoras: Los pacientes pueden presentar debilidad muscular y disminución de reflejos y presentar dificultades para subir y bajar escaleras o para levantarse del suelo. Sin embargo, llevan una vida muy similar a la de la población sin AME.
Esperanza de vida: Estos pacientes tienen una esperanza de vida similar a la de la población sin AME.
Referencias
1. Finkel R, Bertini E, Muntoni F, Mercuri E; ENMC SMA Workshop Study Group. 209th ENMC International Workshop: Outcome Measures and Clinical Trial Readiness in Spinal Muscular Atrophy 7-9 November 2014, Heemskerk, The Netherlands. Neuromuscul Disord. 2015;25(7):593-602.
2. Tizzano EF, Finkel RS. Spinal muscular atrophy: A changing phenotype beyond the clinical trials. Neuromuscul Disord. 2017;27(10):883-889.
3 Grotto S, Cuisset JM, Marret S, et al. Type 0 Spinal Muscular Atrophy: Further Delineation of Prenatal and Postnatal Features in 16 Patients. J Neuromuscul Dis. 2016;3(4):487-495.
4. De Sanctis R, Pane M, Coratti G, et al. Clinical phenotypes and trajectories of disease progression in type 1 spinal muscle atrophy. Neuromuscul Disord. 2018;28(1):24-28.
5. Darras BT. Spinal Muscular Atrophies. In: Clínicas Pediátricas de América del Norte. Elsevier Inc; 2015. p. 743-66.
6. Zerres K, Rudnik-Schöneborn S, Forrest E, et al. A collaborative study on the natural history of childhood and juvenile onset proximal spinal muscle atrophy (type II and III SMA): 569 patients. J Neurol Sci. 1997;146(1):67-72.
7. Faravelli I, Nizzardo M, Comi GP, Corti S. Spinal muscular atrophy--recent therapeutic advances for an old challenge. Nat Rev Neurol. 2015;11(6):351–359.
8. Zerres K, Rudnik-Schöneborn S. Natural history in proximal spinal muscle atrophy. Clinical analysis of 445 patients and suggestions for a modification of existing classifications. Arch Neurol. 1995;52(5):518-523.
9. Mercuri E, Finkel RS, Muntoni F, et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord. 2018;28(2):103-115.
10. Finkel RS, Mercuri E, Meyer OH, et al. Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul Disord. 2018;28(3):197–207.
¿Tienes dudas sobre algún término de este artículo? Consulta el glosario.
#AtrofiaMuscularEspinal #AME #Síntomas #Signos #ManifestacionesClínicas